Capítulo 22
Patología de la glándula suprarrenal
- Dr. Arquímedes Rodríguez C., Dr. Octavio Castillo C.
- Clínica INDISA – Universidad Andrés Bello.
Anatomía e histología de la glándula suprarrenal
Las glándulas suprarrenales son dos órganos retroperitoneales que se encuentran en la región antero-superior y medial de los riñones, incluidos en la grasa perinéfrica. La derecha tiene forma triangular y la izquierda forma semilunar. Su peso aproximado es de 5 g. La porción exterior (corteza), tiene una coloración amarillenta y circunda la porción central (médula) de color gris pálido.1
• Corteza suprarrenal: corresponde al 90% de la glándula y se divide en 3 zonas:
- Glomerular: la más externa. Produce aldosterona (mineralocorticoides). Su regulación depende del sistema renina-angiotensina.
- Fascicular: produce glucocorticoides (cortisol, corticosterona y cortisona) y esteroides sexuales (dehidroepiandrosterona, androstenediona y, en pequeñas cantidades, testosterona, progesterona y estrógenos). Su regulación depende del eje hipotálamo- hipofisario, hormona liberadora de hormona adrenocorticotropa (CRH) y hormona adrenocorticotropa (ACTH).
- Reticular: la más interna, de producción similar a la anterior.
• Médula suprarrenal: sintetiza catecolaminas (adrenalina 80% y noradrenalina 20%).2
Las glándulas suprarrenales están irrigadas por el plexo capsular, compuesto por ramas que proceden de la arteria frénica inferior, la aorta y las arterias renales. Los sinusoides de las zonas fascicular, reticular y de la médula drenan hacia las venas medulares y, por lo tanto, a la vena central. Ésta desemboca, en el lado derecho, a la vena cava inferior y, en el lado izquierdo, a la vena renal.1
| Corteza | Médula | Estroma |
|
Adenoma
-Funcionante (Glucocorticoide/Mineralocorticoide/Virilizante) – No funcionante Carcinoma -Funcionante (más frecuente hipercortisolismo) – No funcionante (40%) |
Ganglioneuroma
Ganglioneuroblastoma Neuroblastoma Paraganglioma Feocromocitoma |
Mielolipoma
Lipoma Fibroma Hemangioma Neurofibroma Linfangioma Sarcoma |
Tabla 1. Tumores Suprarrenales
• Otros:
– Quistes y pseudoquistes.
– Metástasis suprarrenales.
Incidentaloma suprarrenal
El incidentaloma suprarrenal (IS) es definido como una masa suprarrenal, de más de 1cm, encontrado en forma incidental en un estudio radiológico, en ausencia de síntomas o elementos clínicos sugerentes de patología adrenal. El IS es una entidad de causa frecuente en la práctica clínica debido al aumento significativo del número de estudios radiológicos por sintomatología abdominal inespecífica.3,4
La prevalencia de IS como hallazgo en la tomografía computada (TAC) es del 5% y ésta se incrementa con la edad, siendo 0,2% en la tercera década de la vida y 7% en la octava década de la vida. Ocurre con igual frecuencia en hombres y mujeres. La mayoría son adenomas benignos hormonalmente inactivos (82%); sin embargo, existen causas potencialmente letales que deben ser diagnosticadas y tratadas correctamente, como el carcinoma adrenal (4%) y los tumores adrenales funcionantes (11%). Cerca del 3% corresponden a tumores metastáticos, provenientes principalmente de los cánceres de mama, pulmón, riñón o linfoma.3
La evidencia sugiere que los tumores suprarrenales tienen una incidencia de 2 a 5 veces más frecuentes en individuos con diabetes mellitus, obesidad o historia familiar de neoplasia endocrina múltiple (MEN).5,6
Diagnóstico
Clínica
Los tumores funcionantes, benignos o malignos, se caracterizan por la producción excesiva de mineralocorticoides, glucocorticoides, estrógenos, andrógenos y catecolaminas. El porcentaje de masas suprarrenales funcionantes es de un 15 a un 20% según diferentes estudios; un 10% secretan cortisol y originan un síndrome de Cushing, un 5–6% secretan catecolaminas (feocromocitoma) y un 2% secretan aldosterona (hiperaldosteronismo).7,8
El número de masas funcionantes es proporcional al tamaño de la masa. Es muy raro que una masa de menos de 1cm sea funcionante, salvo los pequeños tumores que producen aldosterona, mientras que en el otro extremo el 40% de los tumores mayores de 6cm son funcionantes. La presencia de hiperfunción puede ser inferida por la clínica (palpitaciones, sudoración y cefaleas), por la exploración (hipertensión arterial (HTA), signos típicos del Cushing) o por datos del laboratorio (hipopotasemia).3,4
La ausencia de estos síntomas y signos reduce, pero no excluye la posibilidad de encontrarnos ante un Feocromocitoma, un síndrome de Cushing o un hiperaldosteronismo. En la tabla 2 se resumen los principales signos y síntomas de las patologías que justifican una hiperfunción de la glándula.
| Enfermedad | Síntomas | Signos |
|
Síndrome de Cushing
(10% adenoma funcionante) |
Asintomático si enfermedad subclínica.
Aumento de peso, redistribución centrípeta de la grasa, fascie en luna llena, facilidad hematomas, piel fina y delicada, mala cicatrización, estrías cutáneas vinosas, debilidad muscular proximal, cambios emocionales y cognitivos, infecciones oportunistas, atrofia testicular y ginecomastia en el varón, hirsutismo y acné en la mujer. |
HTA
Osteopenia, osteoporosis Hiperglicemia, diabetes mellitus Hipocalcemia, hiperlipidemias Leucocitosis con linfopenia |
| Feocromocitoma |
Asintomático
Cefalea pulsátil, palpitaciones, sudoración, temblores Taquicardia, dolor abdominal o torácico, debilidad, nauseas, vómitos y anorexia. Crisis espontáneas o desencadenadas por cambios posturales, ansiedad, medicación (metoclopramida, anestésicos), valsalva |
HTA paroxística o constante o ambos
Hipotensión ortostática Palidez Retinopatía I a IV Fiebre |
| Hiperaldosteronismo primario |
Parestesias, debilidad, mialgias.
Cefalea, palpitaciones |
HTA
Hipopotasemia Hipernatremia discreta |
| Carcinoma adrenal |
Dolor abdominal (efecto masa)
Si hipersecreción cortisol asociada (Cushing) Si hipersecreción andrógenos: hirsutismo, acné , amenorrea, aumento libido, piel grasa Si hipersecreción de estrógenos: ginecomastia |
HTA, osteopenia, osteoporosis
Hiperglicemia, diabetes mellitus, hipocalcemia, hiperlipidemias, leucocitosis con leucopenia |
| Metástasis | Historia de tumor extra-adrenal | Signos específicos de tumor primario |
Tabla 2. Signos y síntomas de los principales tumores suprarrenales basado en los datos aportados por Hevia et al.4 HTA= hipertensión arterial
Estudio funcional
Siempre es necesario determinar si el incidentaloma es funcionante o no, lo que puede determinar el diagnóstico de enfermedades que requieren de tratamiento específico, generalmente quirúrgico.
Síndrome de Cushing: resulta de un exceso de cortisol circulante y se caracteriza por la pérdida del ritmo circadiano de esta hormona y una respuesta negativa del eje hipotálamo-hipófisis-suprarrenal. Dependiendo de la cantidad de cortisol que se produzca por el tumor suprarrenal puede haber desde una leve disminución de su ritmo circadiano hasta una atrofia total de la glándula suprarrenal contralateral.3,4,9
La mejor manera para detectar una producción anómala de cortisol es realizar la prueba de supresión con dexametasona (1mg de dexametasona a las 23h y determinación de cortisol a las 8 am del día siguiente). Se han descrito 3 subgrupos de pacientes según niveles de cortisol posterior al test de supresión con 1 mg de dexametasona: normal <50 nmol/L (<1.8 μg/dL), intermedio o dudoso 50-138 nmol/L (1.8–5.0 μg/dL) y francamente alterado >138 nmol/L (>5.0 μg/dL). Se pueden dar falsos positivos si no se realiza bien el test de supresión o por fármacos que aceleran el metabolismo hepático de la dexametasona (anticonvulsivantes).3,4,9,10
Las guías europeas de endocrinología recomiendan realizar pruebas confirmatorias en aquellos pacientes con niveles de cortisol >138 nmol/L (>5.0 μg/dL) posterior a la prueba de supresión con dexametasona.10 El diagnóstico se confirma con la medición plasmática de ACTH, cortisol libre en orina de 24 h, cortisol salival nocturno y un test de supresión con dosis alta de dexametasona durante 2 días (ej. 3 mg, 2 x 2 mg o 8 mg). Así mismo, se recomienda realizar medición plasmática de ACTH en aquellos pacientes con niveles dudosos de secreción autónoma de cortisol 50-138 nmol/L (1.8–5.0 μg/dL) y presencia de comorbilidades (hipertensión arterial, resistencia a la insulina, diabetes mellitus tipo 2, obesidad, dislipidemia y osteoporosis).9,10,11
Síndrome de Cushing subclínico (SCS): En los últimos años ha habido un incremento en el diagnóstico de SCS asociado a incidentaloma suprarrenal, el cual se caracteriza por la hipersecreción autónoma de cortisol (falla a la supresión de cortisol >138 nmol/L (5 μg/dl) posterior a la administración de 1 mg de dexametasona), sin signos clínicos específicos de exceso de cortisol (obesidad central, extremidades delgadas con hipotrofia muscular, cara de luna llena, joroba en la nuca y estrías cutáneas púrpuras, entre otros). Un aumento crónico leve de cortisol endógeno puede tener importantes efectos sistémicos en el cuerpo humano. Se ha descrito una mayor prevalencia de hipertensión arterial, obesidad, alteración de la tolerancia a la glucosa, diabetes mellitus tipo 2, dislipidemia, osteoporosis y eventos cardiovasculares en el SCS.10
Hiperaldosteronismo primario (síndrome de Conn): es un trastorno poco frecuente, que corresponde al 1–2% de todos los incidentalomas.3 Se caracteriza por una producción de aldosterona inadecuadamente alta, relativamente autónoma del sistema renina-angiotensina que no se puede suprimir con una sobrecarga de sodio. Dicha producción inapropiada de aldosterona causa daño cardiovascular, supresión de la renina plasmática, hipertensión, retención de sodio y excreción de potasio que, si es prolongada y severa, puede conducir a hipopotasemia. Puede ser secundario a hiperplasia suprarrenal bilateral (60%), adenoma (35%), hiperplasia suprarrenal unilateral (2%), carcinoma (<1%), tumor ectópico productor de aldosterona (<1%) e hiperaldosteronismo familiar (<1%). La presencia de hipertensión arterial e hipopotasemia debe hacer sospechar la presencia de esta enfermedad. Sin embargo, hoy sabemos que hasta el 70% de los pacientes con hiperaldosteronismo tienen niveles plasmáticos de potasio normales.10,12,13
En aquellos incidentalomas asociados a hipertensión arterial se deberá determinar el cociente aldosterona plasmática [AP (ng/dl)] y actividad de renina plasmática [ARP (ng/ml/h)]. Una AP elevada (>15 ng/dl) con un cociente AP/ARP >30 en dos determinaciones es muy sugerente de una producción autónoma de aldosterona.10,12,14 Un cociente > 100 es diagnóstico y no precisa confirmación. El test de confirmación consiste en la supresión de aldosterona con sobrecarga salina: infusión endovenosa de 2 litros de suero salino en 4 h, entre las 8–12 am, y determinación de AP a las 12 am. Un valor de AP >10 ng/dl confirma el diagnóstico.3 La toma de muestra de aldosterona plasmática mediante cateterismo de la vena suprarrenal bilateral es la prueba estándar de referencia para diferenciar la enfermedad unilateral de la enfermedad bilateral en pacientes con hiperaldosteronismo primario.10,14
Síndrome Adrenogenital: en adultos la virilización en mujeres es un signo de carcinoma productor de andrógenos. En los hombres ginecomastia, atrofia testicular, disfunción eréctil y pérdida de la libido son signos de tumores productores de estrógenos. Éstos son tumores de alto grado, con escasa sobrevida (20% a 3 años). Para la evaluación de este síndrome debemos determinar niveles plasmáticos de dehidroepiandrosterona sulfato (DHEA-S), androstenediona, estradiol y 17-cetosteroides en orina.15
Feocromocitoma: es un tumor neuroendrocrino productor de una o mas catecolaminas (epinefrina, norepinefrina y/o dopamina) derivado de las células cromafines de médula suprarrenal, representa el 5% de los incidentalomas suprarrenales y puede ser esporádico o asociarse a síndromes hereditarios familiares.3 Es importante sospechar, confirmar, localizar, tratar y resecar estos tumores por varias razones. La mayoría de estos tumores hipersecretan catecolaminas y, si no se tratan, el paciente estará expuesto a una alta morbi-mortalidad cardiovascular. Además, los feocromocitomas aumentan de volumen con el tiempo y pueden causar síntomas por efecto de masa e invadir o extenderse a los tejidos y órganos adyacentes. Otra razón que justifica su detección es que, para la enfermedad familiar, la detección de un tumor puede resultar en un diagnóstico y tratamiento más precoz en otros miembros de la familia. Finalmente, algunos feocromocitomas tienen potencial maligno16.
La confirmación diagnóstica se realiza mediante la determinación de metanefrinas libres plasmáticas o metanefrinas fraccionadas en orina. Existe evidencia categórica de que las mediciones de metanefrinas libres en plasma o fraccionadas en orina, son superiores a otras pruebas de exceso de catecolaminas para el diagnóstico de feocromocitoma. Se recomienda que la muestra de sangre para medición de metanefrinas en plasma sea tomada en posición supina, y que el paciente permanezca recostado durante al menos 30 minutos antes del muestreo para evitar falsos negativos.16
A la hora de estudiar y tratar un posible feocromocitoma debemos tener en cuenta las distintas formas de presentación clínica: feocromocitoma bilateral, localización extra-adrenal, feocromocitoma maligno y asociado a endocrinopatía múltiple (MEN).12,15,16,17 El paraganglioma es un tumor derivado de las células cromafines extra-adrenales de los ganglios simpáticos para-vertebrales del tórax, abdomen y pelvis. Los paragangliomas también se pueden originar de ganglios parasimpáticos a lo largo de los nervios vago y glosofaríngeo en el cuello y base del cráneo; estos no producen catecolaminas.16
La presentación familiar asociada a mutación en las subunidades de la succinatodeshidrogenasa (SDH), incluye 5 síndromes de paraganglioma familiar (síndromes PGL del 1 al 5). En la actualidad se recomienda que todos los pacientes portadores de feocromocitoma o paraganglioma participen en la toma de decisiones compartidas para pruebas genéticas. El hecho de que las pruebas genéticas deban considerarse en cada paciente no implica que las pruebas genéticas deben realizarse siempre en cada paciente. En particular, en vista de los costos financieros, las pruebas genéticas tienen un valor limitado en pacientes con feocromocitoma unilateral y sin características del síndrome o malignas y sin antecedentes familiares. La importancia del diagnóstico de una enfermedad hereditaria para las familias en riesgo debe sopesarse frente a los impactos negativos y los costos financieros de las pruebas genéticas.10,16
Un panel de expertos ha propuesto un algoritmo para las pruebas genéticas secuenciales, priorizando la selección de genes a ser probados de acuerdo con una presentación sindrómica o metastásica (Figura 1). El estudio bioquímico adecuado es muy importante para el diagnóstico y manejo de los feocromocitomas/paragangliomas. La determinación del perfil bioquímico (adrenérgico para pacientes con secreción predominante de adrenalina/metanefrina, noradrenérgico para pacientes con secreción predominante de noradrenalina/normetanefrina y dopaminérgico para pacientes con predominio de dopamina/methoxitiramina) puede ser una guía importante para la orientación del estudio genético en ausencia de historia familiar, además de ser marcadores bioquímicos de respuesta al tratamiento y para el seguimiento posterior. Así, las mutaciones en la SDH, en caso de ser secretores, son de perfil más frecuentemente noradrenérgico y/o dopaminérgico.16

Tomado de Guías Clínicas de la Sociedad de Endocrinología: Feocromocitoma y Paraganglioma16
Patología maligna
La posibilidad de malignidad es la principal preocupación del paciente y el médico. El tamaño de la masa y las características radiológicas son los dos principales factores predictivos de malignidad.3
Carcinoma Suprarrenal (CS)
Es un tumor maligno poco frecuente, con una edad media al diagnóstico de 45 años. Los casos localizados son potencialmente curables con cirugía, pero sólo el 30 a 45% de los tumores se encuentra limitado a la glándula suprarrenal al momento del diagnóstico. La sobrevida global a 5 años es del 40%, y los factores pronósticos más importantes son el estadio clínico al momento del diagnóstico según el sistema de clasificación ENSAT10 y la resección completa de la masa tumoral.18 Cerca del 60% de los pacientes presentan síntomas clínicos relacionados con la producción excesiva de hormonas de la corteza suprarrenal. En la tabla 3 se resumen los tipos de presentación clínica del CS.
El CS se asocia en más de la mitad de los casos con hormonas sexuales elevadas y precursores de esteroides. Sin embargo, no se recomienda la medición de estas hormonas en pacientes con incidentalomas suprarrenales de forma rutinaria. En aquellos casos que cursan con una masa suprarrenal indeterminada por imágenes o signos clínicos de exceso de andrógenos, el aumento significativo de hormonas sexuales o sus precursores podría apuntar claramente hacia el CS. Por lo tanto, la medición de dehidroepiandrosterona sulfato (DHEA-S en suero, androstenediona, 17 hidroxiprogesterona, así como testosterona en mujeres y estradiol en hombres y mujeres posmenopáusicas puede demostrar la naturaleza adrenocortical de la masa suprarrenal. Una nueva herramienta muy prometedora para discriminar tumores suprarrenales benignos de malignos aparece en el análisis de un perfil completo de esteroides urinarios medido por cromatografía de gases (GC-MS) o cromatografía de líquidos asociado a masas (LC-MS).10,18
La TAC y la RM son las mejores pruebas para el diagnóstico y estadificación del CS. El 90% de los CS son tumores >4 cm, multilobulados, heterogéneos, con grandes áreas de necrosis y evidencia de invasión vascular. Las metástasis se producen principalmente en el peritoneo, pulmón, hígado y huesos. 19,20
Metástasis suprarrenales
La glándula suprarrenal es el sitio de diseminación metastásica de varios tumores. Cerca del 3% de las masas adrenales son tumores metastáticos, provenientes principalmente de los cánceres de pulmón, riñón, mama, colon o linfoma. Con menor frecuencia se han comunicado casos de metástasis suprarrenal de melanoma, cáncer de próstata e hígado entre otros.3,10,21 La presencia de masas suprarrenales bilaterales aumenta la posibilidad de enfermedad metastásica.10,3. El punto clínico importante es no confundir una lesión metastásica adrenal con un proceso suprarrenal primario. En general, la lesión adrenal es parte del cuadro clínico de la enfermedad metastásica difusa. Se ha comunicado un aumento en la sobrevida de pacientes con tratamiento agresivo del tumor primario pulmonar o renal más adrenalectomía por metástasis suprarrenal.10,21,22,23
Feocromocitoma maligno
Aunque la mayoría de los feocromocitomas/paragangliomas (FC/PG) son benignos, 15 a 17% desarrollan enfermedad metastásica, estando presentes en el diagnóstico inicial en aproximadamente 11 a 31% de los casos. El FC/PG maligno no se puede diagnosticar sobre la base de exámenes histológicos del tumor primario, a pesar del desarrollo de dos sistemas de clasificación histológica diferentes: PASS (Pheochromocytoma of the Adrenal Gland Scaled Score) y GAPP (Grading System for Adrenal Pheochromocytoma and Paraganglioma). Desafortunadamente, se ha comunicado que ambos sistemas de clasificación tienen una alta variabilidad inter-observador y no es adecuado su uso rutinario para distinguir entre tumores benignos y malignos. La malignidad se define como la presencia de metástasis en el tejido no cromafínico como ganglios linfáticos (80%), hueso (72%), hígado (50%), pulmón (50%) y otros. La evolución natural de la enfermedad metastásica es altamente heterogénea, con una tasa de sobrevida global que varía entre 40% y 85% a 5 años de seguimiento.16
Las mutaciones en el gen que codifica la subunidad B de la succinato deshidrogenasa (SDHB) pueden provocar enfermedad metastásica en el 40% o más de los pacientes.10,16 En una serie de la Cleveland Clinic (Cleveland, USA) se describe que el 19% de sus casos fueron malignos, siendo más frecuente este hallazgo en tumores > 5 cm que en los tumores más pequeños (76% versus 24%) y en tumores extra-adrenales (52% versus 9%).24
| 38-40% No funcionante | |
| 50-60% Funcionante | |
| 50-70% | Hipercortisolismo (Síndrome de Cushing) * |
| 20-30% | Exceso de andrógenos (virilizante) en pacientes femeninos* |
| 5% | Exceso de estrógenos (feminizante) en pacientes masculinos* |
| 2-3% |
Exceso de mineralocorticoides Hiperaldosteronismo*
*Frecuentemente combinados |
Tabla 3. Tipos de presentación clínica del carcinoma suprarrenal
Basado en los registros de carcinoma suprarrenal del ENSAT
(European Network for the Study of Adrenal Tumors)18
Características radiológicas de las masas suprarrenales
Tomografía computada (TAC)
La tomografía computada de abdomen es hoy el estudio por de elección para explorar la morfología de la glándula suprarrenal. Permite medir las características de absorción de la estructura en estudio, referidas mediante unidades de absorción denominadas unidades Hounsfield (UH). La caracterización radiológica de una masa suprarrenal como adenoma podría disminuir la necesidad de seguimiento con estudios de imágenes, biopsias y adrenalectomías innecesarias. En ese sentido, se ha descrito que un valor de atenuación <10 UH parece ser un punto de corte seguro en una tomografía computada sin contraste para diferenciar un adenoma/hiperplasia suprarrenal de no-adenomas. Una masa adrenal homogénea, con borde definido y un valor de atenuación <10 UH, es muy probable que se trate de un adenoma benigno.3-6,8-11,12,15, 20, 25
Por otro lado, los adenomas suprarrenales tienen un patrón de lavado del medio de contraste mucho más rápido y se realzan más que los no-adenomas en la TAC. El protocolo de TAC con técnica de lavado (wash-out) incluye la adquisición de imágenes al momento de administrar el contraste, a los 60 segundos y a los 10-15 minutos posterior a la administración del mismo. Las guías europeas de endocrinología establecen que un lavado relativo >40% y un lavado absoluto mayor al 60% sugieren que la masa adrenal es benigna.10 La media de lavado de los adenomas es del 51% a los 5 min y del 70% a los 15 min, frente al 8–20% en lesiones malignas (Tabla 4).3
Resonancia magnética (RM)
Sus indicaciones se limitan al diagnóstico diferencial entre adenomas y metástasis, diagnóstico y localización del feocromocitoma, sospecha de hematoma, trombosis mural y/o signos de invasión de estructuras vecinas en el carcinoma suprarrenal. La RM tiene una sensibilidad y especificidad del 100% para el diagnóstico y extensión del feocromocitoma, dada su característica imagen de hiperintensidad de señal en T2 (imagen de “bombilla” brillante)3,4,24. Además, podría estar indicada en pacientes con FC/PG metastásicos, para la detección de paragangliomas en la base del cráneo y el cuello, en pacientes con clips quirúrgicos que causan artefactos en el TAC, en pacientes con alergia al medio de contraste y en pacientes en los que la exposición a la radiación debe ser limitada (niños, mujeres embarazadas, pacientes con mutaciones de la línea germinal conocidas y aquellas con exposición reciente a radiación excesiva).10,16
Cintigrama suprarrenal
El estudio con 123I-metayodobencilguanidina (MIGB) permite detectar feocromocitomas suprarrenales, paragangliomas o metástasis, debido a que el radionúclido se concentra principalmente en las células procedentes de la cresta neural.3,4,5,9,21. El cintigrama con 123I-MIBG tiene una sensibilidad entre 85-88% para feocromocitomas y entre 56- 75% para los paragangliomas, mientras que la especificidad varía entre 70–100% y 84–100%, respectivamente. Se ha descrito una sensibilidad de 79% para FC/PG metastásicos. Se recomienda la realización de un cintigrama con 123I-MIBG como una modalidad de imagen funcional en pacientes con feocromocitoma o paraganglioma metastásico detectado por otras modalidades de imágenes, cuando se planifica la radioterapia con el isótopo 131I unido a la MIBG, y ocasionalmente en algunos pacientes con mayor riesgo de enfermedad metastásica debido a un tumor primario o extra-suprarrenal de gran volumen (> 6 cm), tumor multifocal (excepto paragangliomas de base de cráneo y cuello) o enfermedad recurrente.10,16
Tomografía con emisión de positrones (PET)
La tomografía por emisión de positrones con 18F-fluorodeoxiglucosa (18F-FDG-PET/CT) es la modalidad de imagen preferida sobre la gammagrafía con 123I-MIBG en pacientes con feocromocitoma/paraganglioma metastásico conocido. Esto debido a que varios estudios han comunicado una superioridad de 18F-FDG PET/CT en comparación con la gammagrafía 131I-MIBG para la detección de FC/PG metastásicos. En general, se demostró que la sensibilidad del PET/CT con 18F-FDG estaba entre 74 y 100%, con el rendimiento más alto para los FC/PG metastásicos, particularmente relacionados con SDHB.10
El descubrimiento de la expresión de receptores de somatostatina en el FC/PG ha promovido el uso de imágenes funcionales (PET/CT) asociadas al péptido 68Ga-DOTA (68Ga-DOTA-TOC, 68Ga-DOTA-NOC y 68Ga-DOTA-TATE). En ese sentido se ha descrito una mayor precisión diagnóstica en el mapeo de FC/PG metastásicos para el 68Ga-DOTATATE PET/CT versus la gammagrafía 131I MIBG y el 18F-FDG PET/CT con una tasa de detección de 93%, 74% y 38%, respectivamente (p < 0,001). Algunos investigadores han sugerido que el 68Ga-DOTATATE PET/CT debería ser considerado como el estudio de imágenes de primera línea en pacientes con sospecha de FC/PG metastásicos ya que al mismo tiempo permitiría la planificación de tratamientos con péptidos análogos de la somatostatina marcados con radionúclidos como el 177Lu-DOTATE y 90Y (Yttrium-90).26
Biopsia por punción con aguja fina
Solo está indicada en caso de sospecha de metástasis, en pacientes con fines de estadiaje de un tumor ya conocido o en aquellos casos donde sospechamos de un carcinoma suprarrenal inoperable, que necesita confirmación histológica para tratamiento oncológico sistémico o inclusión en un ensayo clínico. Siempre se debe excluir previamente la existencia de un feocromocitoma antes de realizar una punción-aspiración con aguja fina, por el peligro de desencadenar una crisis hipertensiva con la punción.4,10,11,20,22,23
| Adenoma | Carcinoma |
|
< 4 cm
Forma: redonda a ovalada – márgenes lisos Densidad homogénea y baja en relación al hígado TAC sin contraste <10 UH TAC con contraste <37 UH a los 30 min Lavado de contraste rápido 40-50% a los 15 min RM señal iso-intensa en T1 y T2 comparada con el hígado |
> 4 cm
Forma: irregular – márgenes mal definidos Densidad heterogénea TAC sin contraste >18 UH TAC con contraste >40UH a los 30 min Lavado de contraste lento RM señal hiper-intensa en T2 comparada con el hígado |
Tabla 4. Características radiológicas de los tumores suprarrenales
TAC: tomografía computada, RM: resonancia magnética, UH: unidades Hounsfield.
Tratamiento
El tratamiento de pacientes portadores de un incidentaloma suprarrenal debe ser discutido por un equipo multidisciplinario con experiencia en tumores suprarrenales (al menos un radiólogo, un endocrinólogo y un urólogo) si presenta uno o mas de los siguientes criterios:
- Estudio de imágenes no categórico para una lesión benigna
- Exceso de secreción hormonal (incluye secreción autónoma de cortisol o Cushing sub-clínico)
- Crecimiento significativo de la masa suprarrenal en el seguimiento con imágenes (crecimiento >20% junto a un incremento de al menos 5 mm en el diámetro mayor)
- Si está considerado el tratamiento quirúrgico
Adicionalmente, el equipo multidisciplinario debe tener acceso a anestesistas y patólogos con experiencia en tumores suprarrenales. La figura 2 muestra un diagrama de flujo general sobre el estudio y manejo del incidentaloma suprarrenal.10
La suprarrenalectomía está indicada en tumores funcionantes y no funcionantes > 4 cm, siendo el estándar de oro la adrenalectomía laparoscópica.23,24 Sin embargo, el abordaje laparoscópico sigue siendo motivo de controversia en el tratamiento del carcinoma suprarrenal, a pesar de que la sobrevida global es similar al abordaje abierto tradicional.21,27
La suprarrenalectomía laparoscópica (SL) se ha usado para la mayoría de las patologías suprarrenales benignas que requieren cirugía. El tiempo de recuperación postoperatoria y la morbilidad a largo plazo asociados a la SL se reducen significativamente cuando esta modalidad se compara con la suprarrenalectomía abierta.27,28 La figura 3 muestra un diagrama de flujo sobre el manejo y vía de abordaje de las masas suprarrenales consideradas a tratamiento quirúrgico.
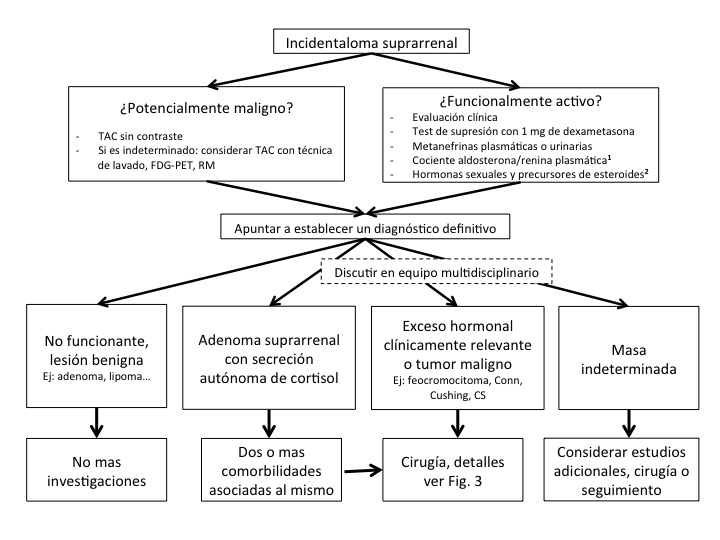
1 En pacientes con hipertensión arterial concomitante y/o hipokalemia
2 En pacientes con características radiológicas sugestivas de carcinoma suprarrenal (CS)
Tomado de Guías Clínicas de la Sociedad Europea de Endocrinología10
Adenoma suprarrenal funcionante
Se recomienda la suprarrenalectomía como tratamiento estándar en adenomas suprarrenales unilaterales con exceso de producción hormonal clínicamente significativa. Se debe discutir la necesidad de suprarrenalectomía en aquellos pacientes con síndrome de Cushing subclínico o secreción autónoma de cortisol, según la presencia o no de comorbilidades asociadas al mismo. Las guías europeas de endocrinología sugieren tratamiento quirúrgico sólo en casos confirmados de secreción autónoma de cortisol que presenten dos o más comorbilidades potencialmente asociadas a hipercortisolismo (HTA, DM2, DLP, osteoporosis), en especial si no pueden ser manejadas adecuadamente con tratamiento médico.10
Masa suprarrenal indeterminada en estudio de imágenes (no funcionante)
Realizar otra modalidad de estudios de imágenes con intervalo de 6-12 meses (TAC con técnica de lavado (wash-out), resonancia magnética o PET/CT) o resección quirúrgica sin mas demora. 10
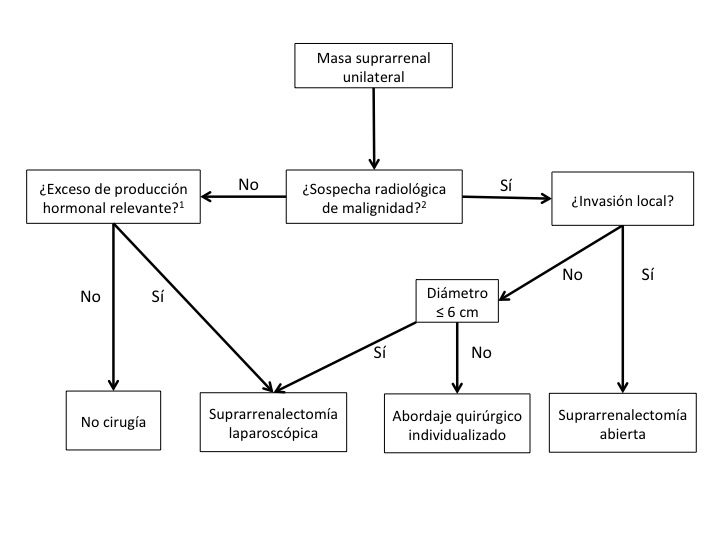
1La secreción autónoma de cortisol no se debe categorizar de forma inmediata como clínicamente relevante.
2Se debe considerar el tratamiento quirúrgico en tumores >4cm con características radiológicas de lesión benigna de forma individualizada.
Tomado de Guías Clínicas de la Sociedad Europea de Endocrinología10
Carcinoma suprarrenal (CS)
La resección quirúrgica completa permanece como la única alternativa potencialmente curativa.10,18,20 La cirugía del CS requiere de cirujanos experimentados en cirugía oncológica y de las glándulas suprarrenales debido a la anatomía específica, el carácter maligno de la enfermedad y la posible necesidad de resección en bloque de múltiples órganos, con el propósito de optimizar la probabilidad de una resección completa (R0) y minimizar el riesgo de complicaciones. La suprarrenalectomía laparoscópica está indicada cuando hay sospecha de CS unilateral <6 cm y sin evidencia de invasión local. Se recomienda un abordaje quirúrgico abierto cuando hay sospecha de CS con compromiso local y/o la masa es ≥6 cm. En casos de invasión de órganos vecinos debe realizarse resección en bloque.
En la actualidad las guías de la sociedad europea de endocrinología recomiendan establecer el diagnóstico de malignidad del tumor suprarrenal utilizando los criterios histopatológicos de Weiss29 (Tabla 5) y proponen la estratificación de los pacientes candidatos a resección completa en dos grupos de riesgo de progresión, tomando en cuenta la combinación entre el sistema de clasificación ENSAT (tabla 6) y el índice de proliferación del marcador tumoral Ki67. De esta manera se definen dos grupos de CS localizado: el CS de riesgo bajo/moderado, que incluye el estadio I-II, R0 y un Ki67 ≤10%, mientras que el CS de alto riesgo incluye el estadio III, R1 o un Ki67 >10%.10,18
| La presencia de tres o más de los siguientes criterios está altamente correlacionada con el comportamiento maligno: |
|
• Alto grado nuclear (criterio de Fuhrman). • Mas de 5 mitosis por 50 campos de alta potencia. • Figuras mitóticas atípicas. • Porcentaje de células claras menor al 25% del total de células tumorales. • Arquitectura difusa (> 33% del tumor). • Necrosis • Invasión venosa (músculo liso en la pared) • Invasión sinusoidal |
Tabla 5. Criterios Histopatológicos de Weiss28
| Estadio ENSAT | Definición |
| I | T1, N0, M0 |
| II | T2, N0, M0 |
| III |
T1-T2, N1, M0
T3-T4, N0-N1, M0 |
| IV | T1-T4, N0-N1, M1 |
Tabla 6. Sistema de clasificación ENSAT10
ENSAT: European Network for the Study of Adrenal Tumors)
T1: tumor ≤ 5cm; T2: tumor > 5cm; T3: infiltración del tejido circundante; T4: invasión de órganos adyacentes o trombo venoso tumoral en vena cava o vena renal; N0: no hay ganglios linfáticos positivos;
N1: ganglios linfáticos positivos; M0: no hay metástasis a distancia; M1: presencia de metástasis a distancia.
Se ha propuesto un diagrama de flujo para el manejo de estos pacientes según la estratificación de riesgo y estadio patológico R0/R1 (Figura 5). Se recomienda tratamiento adyuvante con mitotano para pacientes R0 de alto riesgo y radioterapia externa más mitotano en pacientes R1. En pacientes con CS avanzado al momento del diagnóstico que no califican para el tratamiento local con intención curativa, se recomienda cirugía citorreductora junto al tratamiento con mitotano o mitotano + EDP (Etopósido, Doxorrubicina y Cisplatino), dependiendo de los factores pronósticos (Figura 6).18
De esta manera, factores pronósticos desfavorables como una alta carga tumoral, síntomas no controlados, alto índice proliferativo y evidencia clínica de un tumor de crecimiento rápido, favorecen un enfoque terapéutico activo más agresivo. Además de la cirugía, podemos agregar tratamientos locales como radioterapia, ablación por radiofrecuencia, crioablación, ablación por microondas y quimio-embolización cuando corresponda. Debemos tomar decisiones individualizadas sobre el método de elección basado en la localización de la (s) lesión (es) tumoral (es), la experiencia local, los factores pronósticos y la preferencia del paciente. (Figura 6)10,18
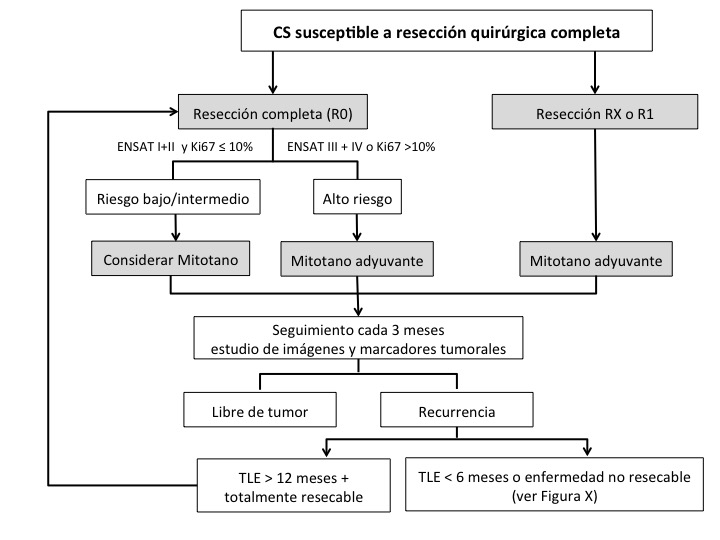
Tomado de Guías Clínicas de la Sociedad Europea de Endocrinología para el manejo del carcinoma suprarrenal18
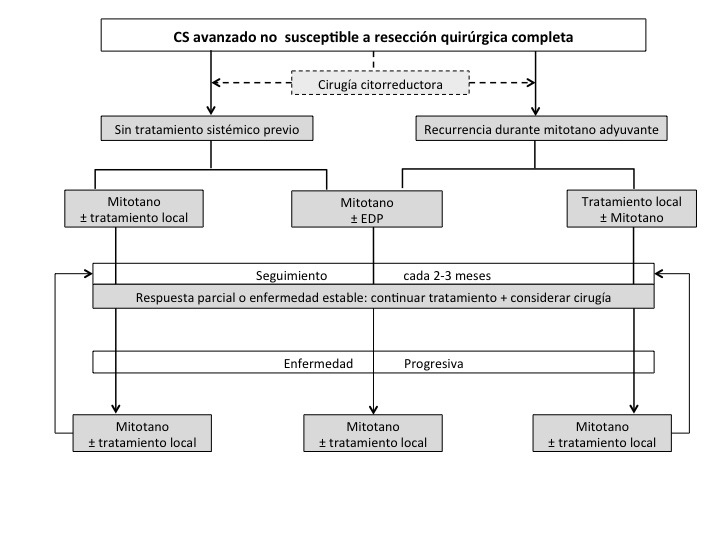
Tomado de Guías Clínicas de la Sociedad Europea de Endocrinología para el manejo del carcinoma suprarrenal18
Feocromocitoma
El tratamiento estándar es la suprarrenalectomía laparoscópica para la mayoría de los feocromocitomas suprarrenales. Se recomienda la resección abierta para feocromocitomas de gran volumen (> 6 cm) o invasivos, para garantizar la resección completa del tumor, prevenir la ruptura del mismo y evitar la recurrencia local. La adrenalectomía parcial está indicada en pacientes seleccionados, como aquellos con feocromocitoma hereditario, con tumores pequeños que ya se han sometido a una suprarrenalectomía contralateral completa, para preservar corteza suprarrenal y prevenir el hipocortisolismo permanente. 10,16,30
El tratamiento quirúrgico del feocromocitoma requiere de un bloqueo preoperatorio para prevenir complicaciones cardiovasculares perioperatorias, siendo la primera opción los bloqueadores de los receptores alfa-adrenérgicos durante 7 a 14 días, tiempo adecuado para normalizar la presión arterial y la frecuencia cardíaca. Los bloqueadores α1-adrenérgicos se han asociado con una presión diastólica preoperatoria más baja, una frecuencia cardíaca intraoperatoria más baja, una mejor recuperación hemodinámica postoperatoria y menos efectos adversos como taquicardia reactiva e hipotensión posoperatoria sostenida que los bloqueadores adrenérgicos no selectivos. 16
Los bloqueadores de los canales de calcio se utilizan como tratamiento complementario para mejorar aún más el control de la presión arterial en pacientes ya tratados con bloqueadores de los receptores α-adrenérgicos. La coadministración preoperatoria de bloqueadores β-adrenérgicos está indicada para controlar la taquicardia, solo después de la administración de bloqueadores α-adrenérgicos. No se recomienda el uso de bloqueadores β-adrenérgicos en ausencia de un bloqueador α-adrenérgico debido a la posibilidad de crisis hipertensivas por la estimulación sin oposición de los receptores α-adrenérgicos.10,16
El tratamiento también debe incluir una dieta alta en sodio y líquidos abundantes para revertir la contracción del volumen sanguíneo inducida por catecolaminas antes de la cirugía y así, prevenir la hipotensión severa después de la extirpación del tumor. En el embarazo el tratamiento es Fenoxibenzamina hasta el parto (cesárea + suprarrenalectomía).4,10,15
Feocromocitoma maligno
El manejo del FC/PG metastásico es un desafío que requiere de un enfoque multidisciplinario: endocrinología, oncología, cirugía, medicina nuclear, radioterapia, radiología intervencionista e histopatología. Hasta ahora, no existe una terapia curativa para pacientes con enfermedad metastásica. Los objetivos del tratamiento son reducir el tamaño del tumor, controlar el exceso de catecolaminas, paliar los síntomas relacionados con la carga tumoral y prevenir la progresión del tumor.30
Las opciones terapéuticas disponibles actualmente son la citorreducción quirúrgica, el tratamiento con radiofármacos, quimioterapia y terapia dirigida. Debe realizarse una resección quirúrgica con resección amplia incluyendo a órganos adyacentes comprometidos. La citorreducción está indicada para disminuir los síntomas del exceso de catecolaminas y para facilitar el manejo médico, incluso cuando no es curativa. Otras opciones de tratamiento paliativo son la radioterapia y la ablación por radiofrecuencia para metástasis óseas dolorosas y embolización arterial, quimioembolización o ablación por radiofrecuencia para metástasis hepáticas. 30
Para el FC/PG metastásico positivo en la cintigrafía con 123I-MIBG y lentamente progresivo, se recomienda el tratamiento con radiofármacos 131I-MIBG como primera línea de tratamiento; siendo el 177Lu-DOTATE y 90Y (Yttrium-90) alternativas potenciales de tratamiento para las personas con metástasis positivas al receptor de somatostatina. La quimioterapia con CVD (Ciclofosfamida, Vincristina y Dacarbazina) y Temozolamida (TMZ) y la terapia dirigida podrían considerarse en caso de enfermedad rápidamente progresiva (Figura 7).30
Las pruebas genéticas de rutina y la medicina personalizada basada en el perfil genómico molecular del tumor conducirán a nuevos objetivos terapéuticos. Varias terapias dirigidas a características únicas del FC/PG se encuentran en diferentes etapas de desarrollo. Actualmente se están llevando a cabo dos ensayos clínicos de fase II de inhibidores del checkpoint (nivolumab, ipilimumab y pembrolizumab) en pacientes con tumores raros, incluidos los FC/PG metastásicos (NCT02834013, NCT02721732).30
Neuroblastoma, ganglioneuroblastoma y ganglioneuroma
El tratamiento en los estadios I y II es la resección quirúrgica asociada a quimioterapia con tasas de sobrevida del 80%. En estadios más avanzados se realiza radioterapia + quimioterapia previo a la cirugía.
Quistes suprarrenales
Los más frecuentes son los quistes endoteliales o linfangiectásicos (45%) y los pseudoquistes (39%). Si son asintomáticos, homogéneos y sin crecimiento deben ser controlados tras punción y análisis del líquido, pero si son sintomáticos, >3,5 cm, o con material hemorrágico deben drenarse percutáneamente o por laparoscopia.15
A pesar de que, en la mayoría de los casos, los pacientes son derivados de distintas unidades con el diagnóstico hecho y con todos los estudios realizados, creemos que el urólogo debe tener presente los conocimientos básicos en el manejo de masas suprarrenales. En este sentido, existen muchos urólogos que realizan su actividad diaria en centros donde no disponen de servicio de endocrinología, siendo necesario en estos casos conocer los principios básicos en el manejo del incidentaloma suprarrenal.
Actualmente no cabe duda sobre la necesidad de la suprarrenalectomía en masas adrenales funcionantes ni en procesos con sospecha de malignidad; siendo el estándar dorado la cirugía laparoscópica

Bibliografía
1.- McNicol AM. The human adrenal gland. Aspects of structure, function and pathology. In: James VHT, editor. The adrenal gland. 2nd ed. New York: Raven Press; 1992 p. 1 – 42.
2.- Fraser R. Biosynthesis of adrenocortical steroids. In: James VHT (ed). The adrenal gland. 2nd ed. New York: Raven Press; 1992 p. 117 – 130.
3.- Young WF Jr. Management approaches to adrenal incidentalomas. A view from Rochester, Minnesota. Endocrinol Metab Clin North Am. 2000;29:159-85.
4.- Hevia M, Abascal JM, Boix P, Dieguez M, Delgado E, Abascal JM et al. [Management of adrenal mass: What urologists should know]. Actas Urol Esp. 2010;34:586-91. Review
5.- Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev. 2004;25:309-40. Review.
6.- Management of the clinically inapparent adrenal mass (incidentaloma). National Institutes of Health (NIH) State-of-the-Science Conference Statement. 2002, February 4-6.
7.- Mantero F, Masini AM, Opocher G, Giovagnetti M, Arnaldi G. Adrenal incidentaloma: an overview of hormonal data from the National Italian Study Group. Horm Res. 1997;47:284-9.
8.- Bülow B, Jansson S, Juhlin C, Steen L, Thorén M, Wahrenberg H et al. Adrenal incidentaloma – follow-up results from a Swedish prospective study. Eur J Endocrinol. 2006;154:419-23.
9.- Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-10. Review.
10.- Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2016;175:G1-G34.
11.- Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Alì A et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85:637-44.
12.- Li-Ming SU. Adrenal Mass: Contemporary Evaluation and Management. AUA Annual Meeting Course 026 PG. 2014, May 16-21.
13.- Castillo OA, Díaz M, Arellano L. [Partial laparoscopic adrenalectomy in primary hyperaldosteronism]. Actas Urol Esp. 2011;35:119-22.
14.- Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M et al. Endocrine Society. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:3266-81.
15.- Broseta E, Budía A, Burgués J.P, Luján S, Jiménez Cruz J.F. Tumores suprarrenales. En: Urología Práctica 2011. Valencia: pp 218 – 220.
16.- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH et al. Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915-42.
17.- Castillo OA, Campos R, Henríquez R, Bravo J. Adrenalectomía laparoscópica bilateral sincrónica en feocromocitoma bilateral. Rev Chil Cir. 2011; 63: 573-78.
18.- Fassnacht M, Dekkers OM, Else T, Baudin E, Berruti A, de Krijger R et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2018;179:G1-G46.
19.- Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol. 2003;169:5-11. Review.
20.- Sepúlveda F, Saldías R, Castillo OA. Carcinoma Suprarrenal. Rev. chil. Urol. 2009; 74: 9-14.
21.- Castillo OA, Vitagliano G, Kerkebe M, Parma P, Pinto I, Diaz M. Laparoscopic adrenalectomy for suspected metastasis of adrenal glands: our experience. Urology. 2007;69:637-41.
22.- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol.
2000;143:91-7.
23.- Kasperlik-Zeluska AA, Rosłonowska E, Słowinska Srzednicka J, Migdalska B, Jeske W, Makowska A et al. Incidentally discovered adrenal mass (incidentaloma): investigation and management of 208 patients. Clin Endocrinol (Oxf). 1997;46:29-37.
24.- Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev. 2003;24:539-53.
25.- Castillo OA, Sánchez-Salas R, Vidal I. Laparoscopic adrenalectomy. Minerva Urol Nefrol. 2008;60:177-84. Review.
26.- Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of(68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J Nucl Med. 2019;60:369-376.
27.- Castillo OA, Rodríguez-Carlin A, López-Vallejo J, Borgna V. Complications associated with laparoscopic adrenalectomy: Description and standardized assessment. Actas Urol Esp. 2014;38:445-50.
28.- Castillo OA, Vitagliano G, Secin FP, Kerkebe M, Arellano L. Laparoscopic adrenalectomy for adrenal masses: does size matter? Urology. 2008;71:1138-41.
29.- Weiss LM, Medeiros LJ, Vickery AL Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol. 1989;13:202-6.
30.- Corssmit EPM, Snel M, Kapiteijn E. Malignant pheochromocytoma and paraganglioma: management options. Curr Opin Oncol. 2020;32:20-26.
